28F G9P1A7 currently 8 weeks pregnant presented to the ED as a transfer from outside hospital for suspected ectopic. Patient reports 6/10 generalized abdominal pain and took Acetaminophen with minimal relief. States that she has been following with outside hospital for her pregnancy and was found to have inappropriately rising HCG levels. Reports vaginal bleeding x3 weeks with irregular menstrual periods. Patient denies fever, chills, nausea, vomiting, chest pain, shortness of breath, dizziness, weakness, calf pain.
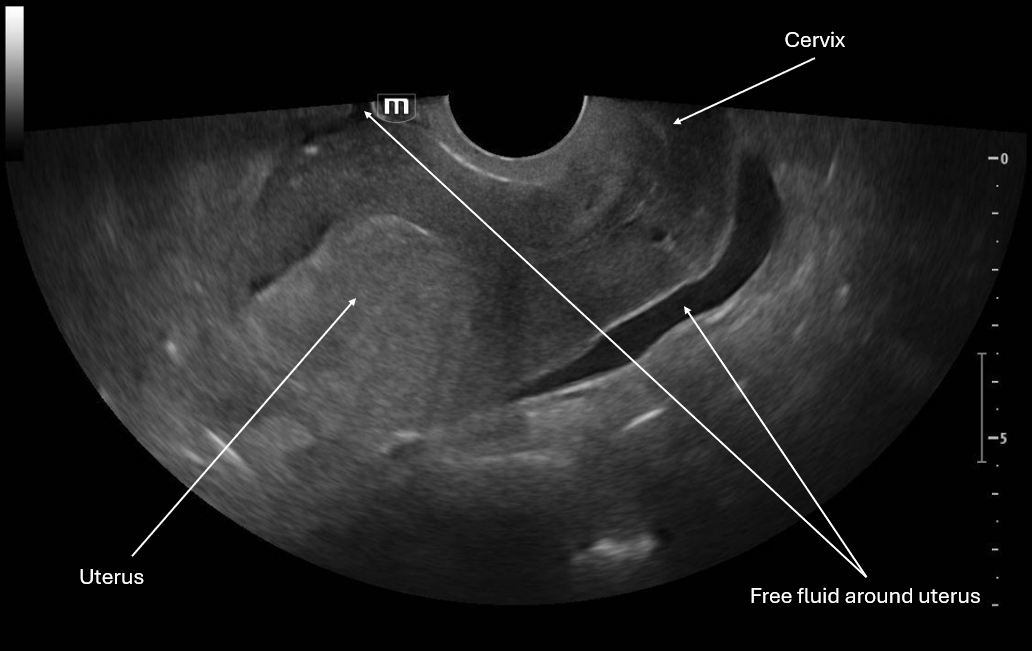
This image is a sagittal view of the uterus with the endocavitary probe marker directed up. From this we can see the fundus of the uterus and cervix with some pelvic free fluid extending past a third of the uterus.
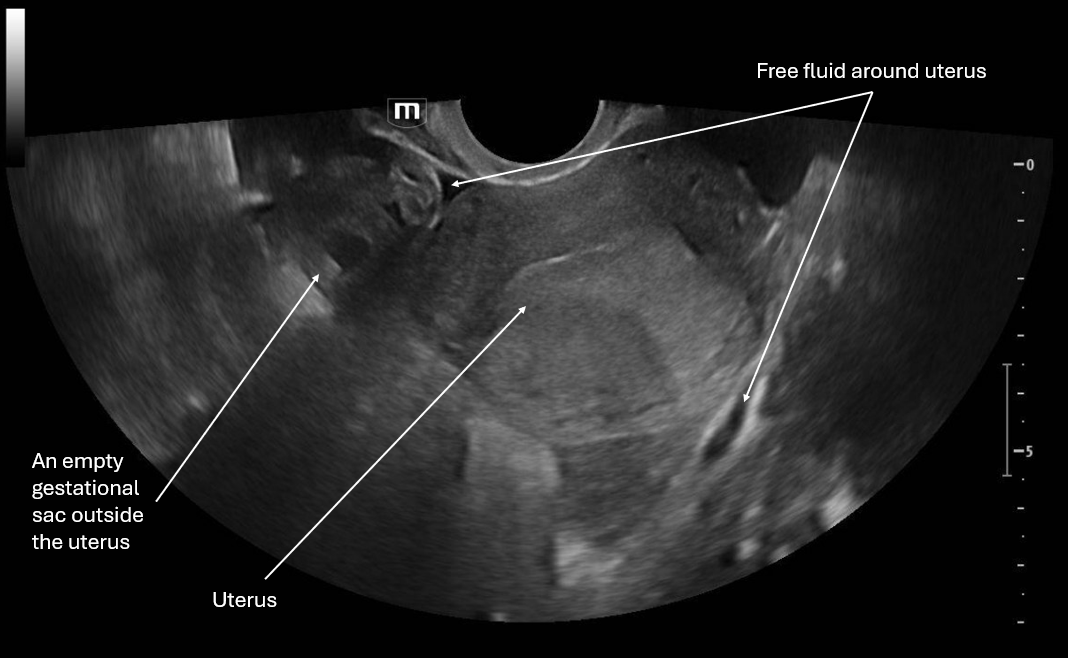
This image is a transverse view of the uterus where the probe marker is towards the patient's right. There is the fundus of the uterus in the middle of the screen and on the patient's right there is what appears to be an empty gestational sac and again we see pelvic free fluid.
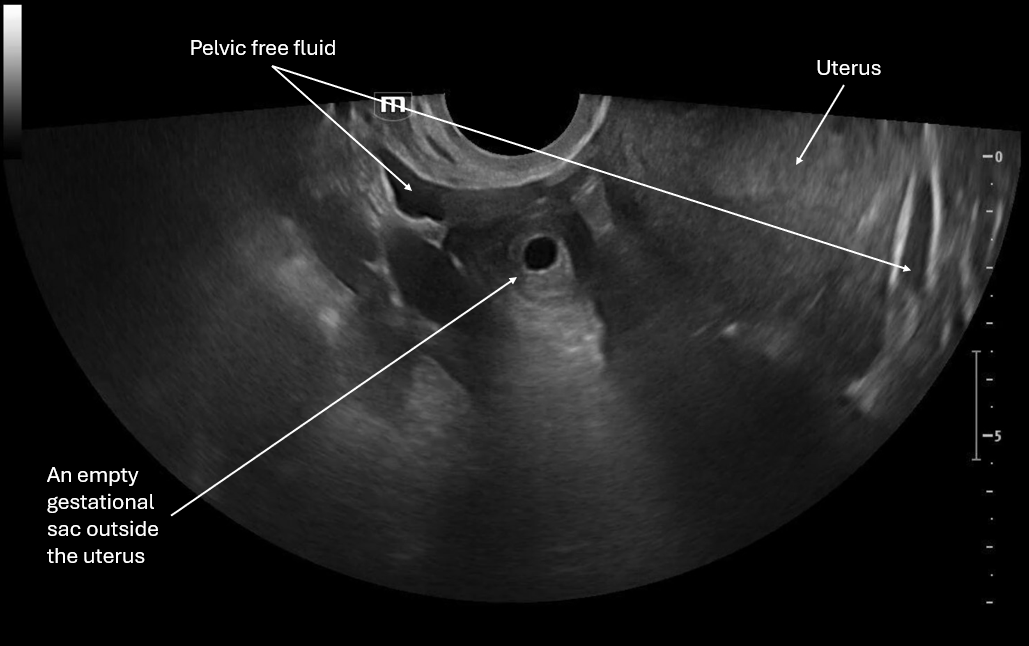
This image focuses on what appears to be an empty gestational sac outside the uterus and we again see pelvic free fluid.
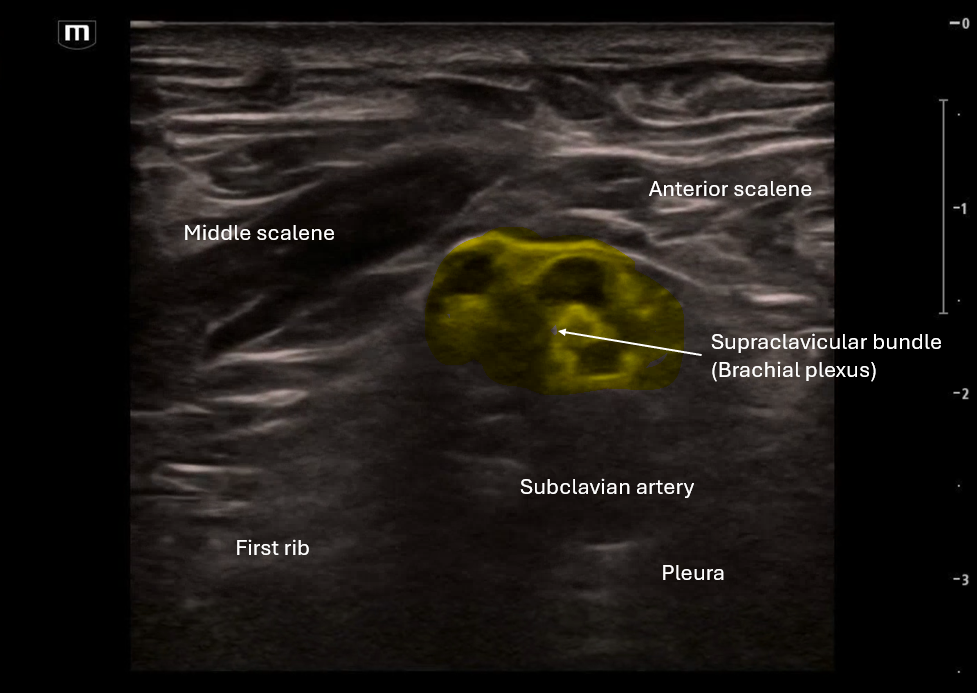
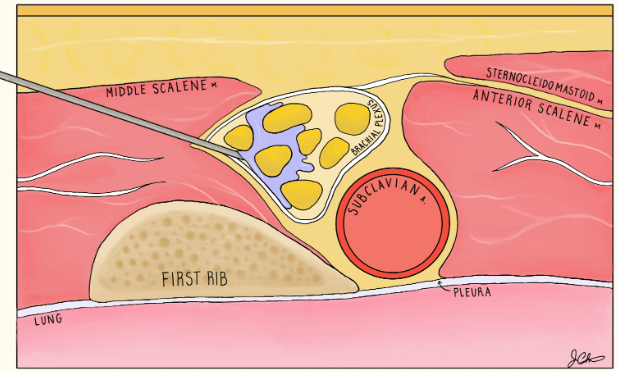
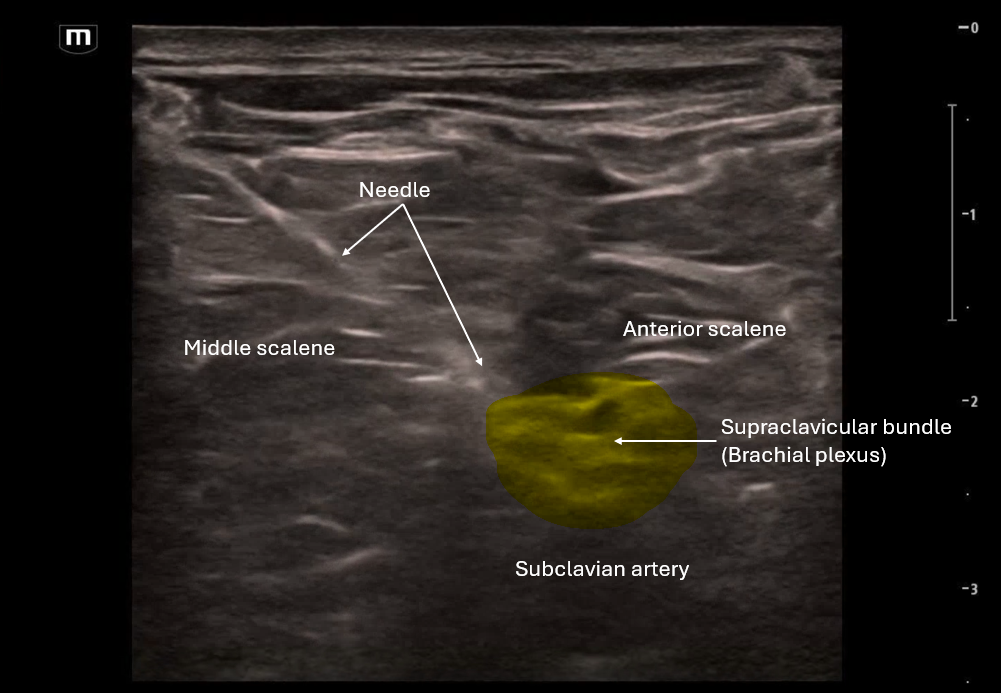
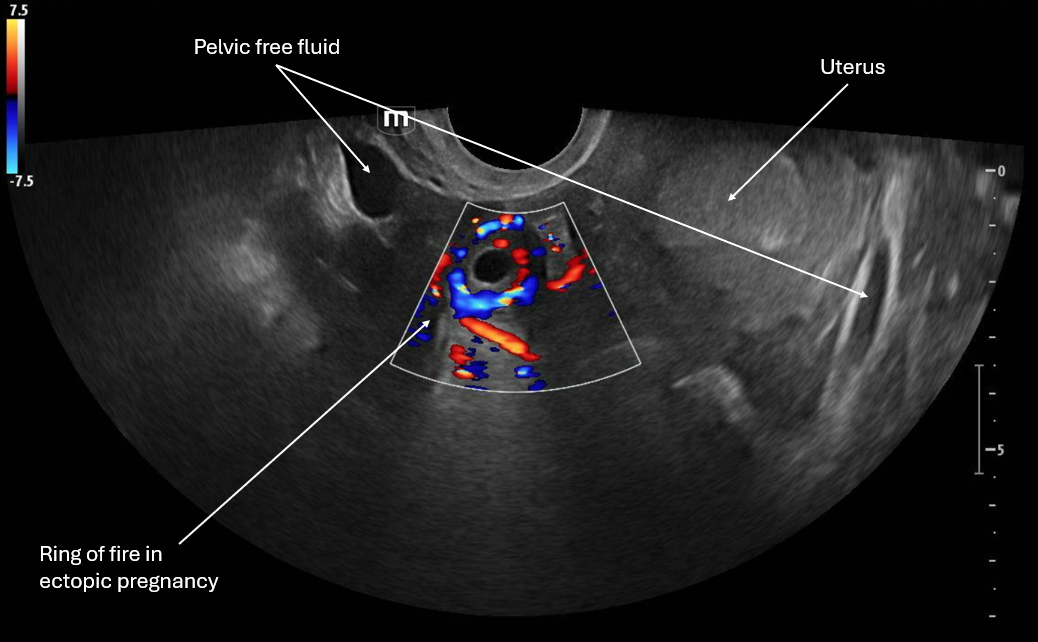
This image applies color flow to the suspected ectopic pregnancy and this shows the "ring of fire" sign which indicates an area of high blood flow due to increased vascularity to the ectopic pregnancy. This "ring of fire" can also be a normal finding with corpus luteum cysts so correlate with other ultrasound findings like free fluid in the pelvis and other elements of the clinical picture.
Patient was taken to the OR and a right laprascopic salpingectomy was performed for a right tubal ectopic pregnancy with about 100cc of blood in the pelvis. Patient was discharged.
POCUS Pearls for Ectopic Pregnancy and "Ring of Fire" sign
Circumferential Doppler flow around adnexal mass = hypervascular tissue
NOT diagnostic alone (corpus luteum can look identical)
Think ectopic if: +hCG + empty uterus + adnexal mass
Free fluid in the pelvis, RUQ +/- unstable vital signs is a ruptured ectopic until proven otherwise